Technical Aspects of DFT Calculations
Grid Size Selection
Evaluation of the exchange-correlation energies of all density functional methods implemented in Gaussian involves a grid-based numerical integration step. The computational effort required for this step strongly depends on the selected grid size. The larger the number of integration points per atom, the higher is the computational cost and the better the numerical accuracy of the calculation. Several predefined grids are available in Gaussian, which can be used through the keyword
int=(gridsize) or int=(Grid=gridsize)
Alternatively, the grid size can also be specified with IOP settings, using either IOP(5/44=gridsize) or IOP(3/75=gridsize), depending on the version of Gaussian. The following example shows, how the numerically accurate ultrafine grid can be used in a frequency calculation of the methanol-1-yl radical at the UB3LYP/6-31G(d) level of theory:
#P UB3LYP/6-31G(d) freq iop(3/75=5)
UBecke-3-LYP/6-31G(d) freq methanol-1-yl radical
0 2
C1
O2 1 r2
H3 2 r3 1 a3
H4 1 r4 2 a4 3 d4
H5 1 r5 2 a5 4 d5
r2=1.37007444
r3=0.96903862
r4=1.08886325
r5=1.08381987
a3=108.87657926
a4=118.49962766
a5=112.61039767
d4=-29.19863769
d5=-146.73450089
|
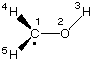 |
Using various settings for gridsize the following results are obtained for the system shown above:
| gridsize | corresponding IOP setting |
(radial shells * angular points) |
CPU [sec] | Etot | low frequencies (cm-1) |
|---|---|---|---|---|---|
| CoarseGrid | 2 | (35*110), pruned | 34 | -115.0520127 | -110.9325 -0.0015 -0.0009 -0.0008 24.6825 59.6546 |
| SG1Grid | 1 | (50*194), pruned | 45 | -115.0520371 | -30.5802 0.0015 0.0015 0.0016 10.5877 29.2740 |
| finegrid | 4 | (75*302), pruned | 84 | -115.0520326 | -20.7294 -8.3913 0.0009 0.0009 0.0014 11.1426 |
| ultrafine | 5 | (99*590), pruned | 215 | -115.0520324 | -0.0013 -0.0013 0.0011 2.6708 4.0642 14.0308 |
| 099590 | 099590 | (99*590), not pruned | 909 | -115.0520324 | -0.0012 -0.0010 -0.0008 3.0995 4.5225 14.0401 |
| mmmnnn | mmmnnn | (mmm*nnn), not pruned | - | - | - |
The CPU times (in sec.) and total energies listed here together with the grid specifications are those for the UB3LYP/6-31G(d) frequency calculation on the methanol radical on the CIP room Pentium 4 LINUX PCs. It is clear from these results that neither the total electronic energy nor the low-frequency vibrations converge quickly with increasing grid size. Thus, for each application it must be decided, whether the substantial computational effort for a very high grid size is justified. It is also important to note that the energy variation with grid size is significant enough that all calculations for stationary points on the same potential energy surface must be performed with the same grid in order to be comparable!
In addition to the grid selection for integral evaluation using the int=(gridsize) keyword, a particular grid can also be selected for the calculation of second derivatives using the CPHF=gridsize keyword. The available options for gridsize are identical in both cases. If no explicit grid is specified through the CPHF keyword, the following relations exist between the grids used for integral and second derivative calculations:
| gridsize used for integral evaluation |
grid used for second derivatives |
|---|---|
| CoarseGrid | CoarseGrid |
| SG1Grid | CoarseGrid |
| finegrid | CoarseGrid |
| ultrafine | SG1Grid |
| (mmm*nnn) | (mmm*nnn) |
These combinations are usually appropriate for most systems. The default grid size for integral evaluation in the most recent versions of Gaussian is finegrid. Better grids are required for kinetic isotope effect calculations, the optimization of stationary points on very flat potential energy surfaces, and IRC-following on flat potential energy surfaces. Increasing the CPHF grid size over what is specified per default through the integral grid selection is rarely helpful. Smaller grids can be quite useful to speed up geometry optimizations of large systems, especially when still far away from the next stationary point.
Treating open shell systems
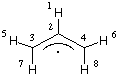
For practical reasons the DFT methods also use a DFT "wavefunction" constructed as an antisymmetrized product of one electron functions (now termed Kohn-Sham orbitals). As in Hartree-Fock theory, electrons of unlike spin can either use the same spatial orbitals (restricted DFT methods) or different spatial orbitals (unrestricted DFT methods). Specification of a restricted open shell wavefunction for the Becke3LYP hybrid functional requires the ROB3LYP keyword, while the unrestricted version is specified using UB3LYP. If no explicit choice is made in the input file, open shell systems are treated with the unrestricted approach by default. It should be noted that unrestricted DFT methods (GGA or hybrid functionals alike) are much less affected by the phenomenon of spin contamination than Hartree-Fock theory and also give much better predictions of EPR spectra of organic radicals. The UB3LYP/6-31G(d) calculation of the allyl radical is achieved with the following input file:
#UBecke3LYP/6-31G(d) scf=(tight)
UB3LYP/6-31G(d) ally radical (C2v)
0 2
H
C,1,r2
C,2,r3,1,a3
C,2,r3,1,a3,3,180.
H,3,r5,2,a5,1,0.
H,4,r5,2,a5,1,0.
H,3,r7,2,a7,1,180.
H,4,r7,2,a7,1,180.
r2=1.09054601
r3=1.38611149
r5=1.08492921
r7=1.08690606
a3=117.45043254
a5=121.64506508
a7=121.12827199
|
 |
Comparison of the expectation value of the <S2> operator calculated at the UHF/6-31G(d) level (0.9729) with that obtained at UB3LYP/6-31G(d) (0.7818) shows that the spin contamination problem is much reduced with DFT methods. From the construction principle of the hybrid DFT methods it is clear that a larger HF-exchange component will also give rise to larger amounts of spin contamination. Using the allyl radical at the UB3LYP/6-31G(d) structure as an example, the <S2> value amounts to 0.7650 at the BLYP/6-31G(d) level and to 0.8228 at the BHandHLYP/6-31G(d) level. The precise values of the expectation value of the <S2> operator will also depend to some extend on the basis set used.
Another important point to consider in DFT calculations concerns the self interaction error resulting from an artificial interaction of the electron density with itself. While the definition of the exchange and Coulomb energies in Hartree-Fock theory leads to a perfect cancellation of these self interaction terms, the cancellation is incomplete in DFT methods. As nicely explained by Koch and Holthausen in their monograph on DFT methods, this can be easily demonstrated using the hydrogen atom. The exact energy for this one-electron system is -0.50000 Hartree. As there is no second electron in the system, no correlation energies have to be calculated and Hartree-Fock theory is able to provide the exact answer in the complete basis set limit. Using a basis set considered "large" for molecular systems such as the cc-pVQZ basis set, a value of -0.499946 Hartree is indeed obtained. Due to the parameterized exchange and correlation functionals, however, much more negative values can be obtained in particular with hybrid DFT functionals. Using the same basis set, the UB3LYP functional predicts a value of -0.502346 Hartree!!
last changes: 06.02.2008, HZ questions & comments to: zipse@cup.uni-muenchen.de