Complete Basis Set (CBS) Extrapolation Schemes
Complete Basis Set (CBS) extrapolation schemes are used to extrapolate results obtained in quantum mechanical calculations with finite basis sets to the basis set limit. The details of these extrapolation schemes depend on the particular quantum mechanics used as well as the (finite) basis sets employed. Many CBS extrapolation schemes are based on the correlation consistent basis sets developed by Dunning and coworkers (commonly abbreviated as cc-pVnZ, where the "cardinal number" n = D, T, Q, 5, 6 etc.) as one of the first systematically developed basis set families.[1] Basis set extrapolations are most useful (and also most important) for highly correlated electronic structure methods, where basis set effects are large and often quite systematic in nature, and where calculations with large basis sets are often prohibitively expensive. Taking CCSD(T) single point calculations for the water molecule as an example, we obtain the electronic energies Etot listed in Table 1 for different members of the cc-pVnZ basis sets. These energies are calculated in the following sequential steps: a) Calculation of the Hartee-Fock energy ESCF (for closed shell systems such as water, this defaults to the Restricted Hartree Fock (RHF) and for open shell system to the Unrestricted Hartree Fock (UHF) or the Restricted Open Shell Hartree Fock (ROHF) methods). This energy is sometimes also referred to as the "reference energy". b) Based on the SCF orbitals the correlation energy is then calculated with the iterative CCSD method (Ecorr,CCSD). c) The correlation energy due to triple excitations (Ecorr,T) is calculated next using a method based on perturbation theory. d) Finally, the two correlation energies are summed up to yield the overall correlation energy (Ecorr) and added to the reference energy ESCF to yield the combined Etot at CCSD(T) level. In order to reduce the computational effort, most CCSD(T) calculations employ the "frozen core (FC)" approximation, where only the valence electrons are included in the correlation energy calculations. For the example of water presented in Table 1, the correlation calculations thus include only eight of the ten electrons present in the system. An input file for this type of calculation with ORCA can be found HERE and with Gaussian can be found HERE.
Table 1. CCSD(T,FC)/cc-pVnZ energies [in Hartree] for water in its B3LYP-D3/6-31G(d) geometry (calculations have been performed with ORCA; the frozen core approximation has been used in all cases).
| basis set | cardinal number n |
ESCF | Ecorr | Etot |
| cc-pVDZ | 2 | -76.02601029 | -0.215194298 | -76.241204592 |
| cc-pVTZ | 3 | -76.05609404 | -0.275956872 | -76.332050910 |
| cc-pVQZ | 4 | -76.06370993 | -0.295870929 | -76.359580863 |
| cc-pV5Z | 5 | -76.06596062 | -0.302868791 | -76.368829406 |
| cc-pV6Z | 6 | -76.06627709 | -0.305534810 | -76.371811902 |
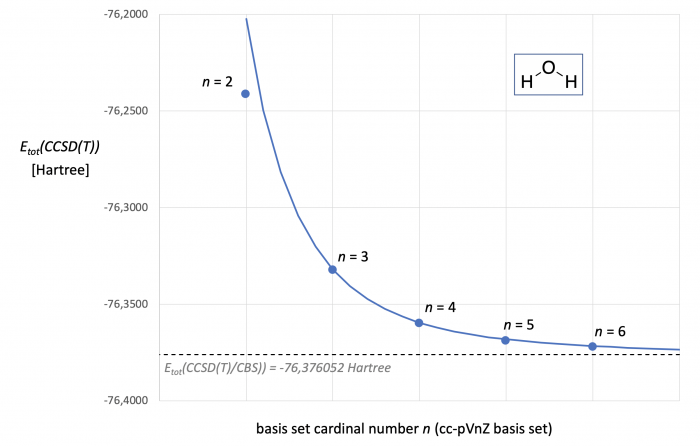
Figure 1. CCSD(T,FC)/cc-pVnZ energies [in Hartree] for water as a function of basis set size. The dotted line marks the Etot(CCSD(T)/CBS) energy predicted through two point extrapolation with the cc-pVTZ and cc-pVQZ basis sets. The blue curve represents Etot(CCSD(T)) as a continous function of cardinal number n.
Ploting the total energies (Etot) for water obtained at the CCSD(T) level against the basis set cardinal number n as in Fig. 1 suggests a systematic (most likely asymptotic) relationship. However, Hartree-Fock energies and correlation energies show systematically different dependencies on basis set size, and most extrapolation schemes thus employ different extrapolation equations for ESCF and Ecorr. A frequently used extrapolation scheme for CCSD(T) calculations goes back to work of Helgaker et al. and Petersson et al.[2,3] The SCF energy ESCF(n) for a basis set with cardinal number n is in this case given through eq. (1). The SCF energy at the CBS limit (ESCF(CBS)) can thus be obtained through a two-point extrapolation with results from calculations with cardinal numbers n and the next higher cardinal number m according to eq. (2). While for specifically optimized basis sets a value of z = 6.30 has been found to work well,[3] optimized z-values have been reported for specific n/m pairs and basis set families. For the cc-pVnZ basis set family, for example, z = 5.4 has been proposed for n = 3/m = 4 extrapolations.[4] Using these latter values we obtain for water ESCF(CBS) = -76.0660048 Hartree. Combination of this result with all data for n = 4 yields A = 126.8299023 in eq. (1).
Correlation energies show an exponential dependence on the basis set cardinal number n as expressed by eq. (3). The corresponding two-point extrapolation with basis sets of cardinal numbers n and m employs eq. (4). The exponent y is here expected to assume values close to y = 3, but the optimum choice may again depend on the basis set family and the n/m values employed.[4] For the cc-pVnZ basis set family an optimum value of y = 3.05 has been found for the 3/4 extrapolation. This type of calculation can actually be performed in a fully automated way in ORCA as shown HERE. For the correlation energies listed in Table 1a for water we obtain Ecorr(CBS) = -0.310047536 Hartree und thus Etot(CCSD(T)/CBS) = -76.3760523 Hartreee (shown as a dotted line in. Fig. 1). Inserting this result into eq. (3) with data for n = 4 yields A = 0.9724231. With the A, z, and y values for eqs. (1) and (3) in hand we can predict Etot(CCSD(T)/cc-pVnZ) as a continous function of n. Comparison of these energies (shown in Fig. 1 as the grey curve) with energies calculated for discrete cardinal numbers (blue points in Fig. 1) shows very good aggreement for all values of n, except for n = 2. It is indeed found also for other basis set families that CBS extrapolations with double-zeta qualitiy basis set are less reliable and require different choices of z- and y-parameters as compared to extrapolations with higher cardinal numbers.
The enormous computational demands of CCSD(T) calculations and their unfavorable scaling behavior with system size puts severe limits to CCSD(T)-based CBS extrapolations with higher basis sets. The Domain-Based Local Pair Natural Orbital-(or DLPNO)-CCSD(T) method is one of the fastest currently available approximate CCSD(T) methods that scales linearly with system size.[4] For water as a model system the respective Hartree-Fock and correlation energies calculated at DLPNO-CCSD(T) level are shown in Table 2. Matching these results with those in Table 1 illustrates that the ESCF energies obtained here are practically identical, while more notable differences exist in the correlation energies (and thus also the final Etot(DLPNO-CCSD(T)) values). CBS extrapolation using the same expressions as before for the canonical CCSD(T) calculations and the results obtained with the cc-pVTZ and cc-pVQZ basis sets yield ESCF(CBS) = -76.0660046, EDLPNO-corr(CBS) = -0.3098850, and thus Etot(DLPNO-CCSD(T)/CBS) = -76.375890 Hartree (shown as a dotted line in Fig. 2). With these results we can again use eqs. (1) and (3) to predict Etot(DLPNO-CCSD(T)/cc-pVnZ) as a continous function of n (green curve in Fig. 2). As already seen in the canonical CCSD(T) calculations, all results obtained with the cc-pVnZ basis set family except for cc-pVDZ fall nicely on this line.
Table 2. DLPNO-CCSD(T,FC)/cc-pVnZ energies [in Hartree] for water in its B3LYP-D3/6-31G(d) geometry (calculations have been performed with ORCA; the frozen core approximation has been used in all cases).
| basis set | cardinal n |
ESCF | Ecorr | Etot |
| cc-pVDZ | 2 | -76.02601029 | <-0.215221297 | -76.241231591 |
| cc-pVTZ | 3 | -76.05609404 | -0.275869676 | -76.331963714 |
| cc-pVQZ | 4 | -76.06370993 | -0.295739738 | -76.359449672 |
| cc-pV5Z | 5 | -76.06596062 | -0.302730784 | -76.368691400 |
| cc-pV6Z | 6 | -76.06627709 | -0.305476138 | -76.371753231 |
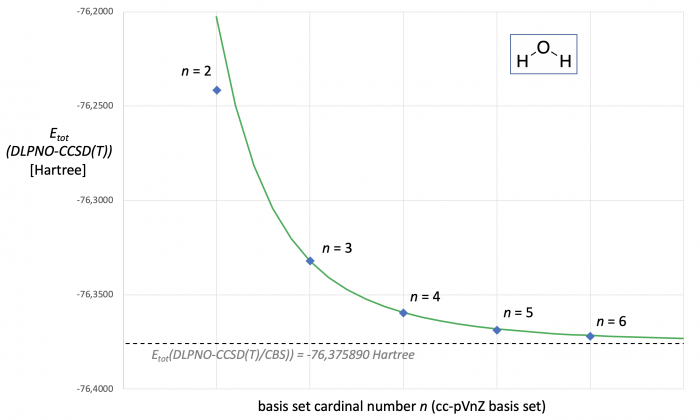
Figure 2. DLPNO-CCSD(T,FC)/cc-pVnZ energies [in Hartree] for water as a function of basis set size. The dotted line marks the Etot(DLPNO-CCSD(T)/CBS) energy predicted through two point extrapolation with the cc-pVTZ and cc-pVQZ basis sets. The green curve represents Etot(DLPNO-CCSD(T)) as a continous function of cardinal number n.
It may be technically meaningful to judge the quality of approximate CCSD(T) methods by the fraction of the correlation energy recovered as compared to the canonical CCSD(T) reference (either for a discrete basis set or for the CBS limit). For computational chemistry applications it is, however, much more relevant to compare reaction and activation energies predicted with these two approaches. To this end we analyze here the formal hydrogen transfer reaction between hydroxyl radical (1) with methanol to yield the methoxy radical (2) and water. The reaction enthalpy of this transformation shown in eq. (5) is sometimes referred to as the radical stabilization energy (RSE) of the methoxy radical (2). Calculations of this latter species in the Cs point group are complicated by the fact that two close lying electronic states of similar energy exist. For the theoretical methods used here the 2A' state is of lower energy and has therefore been used in the RSE(2) calculations in Table 3. An input file for this calculation can be found HERE. Experimental heat of formation data is available for all four species in equation (5), and the resulting reaction enthalpy of this transformation amounts to RSE(2,exp) = -57.0±0.3 kJ/mol. From the results in Table 3 we first of all see that the CCSD(T) and DLPNO-CCSD(T) results differ by no more than 0.8 kJ/mol, whatever the choice of basis set or extrapolation scheme. We furthermore see that both CBS extrapolation schemes as well as calculations with the cc-pV5Z basis set are within 1 kJ/mol of the experimental result. Largest deviations are, as expected, observed for the cc-pVDZ basis set.
Table 3. Radical stabilization energy of methoxy radical 2 (RSE(2)) calculated at various levels of theory.
| basis set | cardinal number n |
RSE(2) (CCSD(T))[a,b] |
RSE(2) (DLPNO-CCSD(T))[a,b] |
| cc-pVDZ | 2 | -47.6 | -48.0 |
| cc-pVTZ | 3 | -53.0 | -52.8 |
| cc-pVQZ | 4 | -55.4 | -55.0 |
| cc-pV5Z | 5 | -56.6 | -56.0 |
| CBS(3/4)[c] | -56.8 | -56.1 | |
| CBS(4/5)[d] | -57.5 | -56.7 | |
| exp.[e] | -57.0±0.3 | -57.0±0.3 |
[a] The frozen core approximation has been used. [b] (U)B3LYP-D3/6-31G(d) geometries have been used in all cases. [c] Two-point extrapolation with the cc-pVTZ and cc-pVQZ basis sets, z = 5.40 and y = 3.05. [d] Two-point extrapolation with the cc-pVQZ and cc-pV5Z basis sets, z = 6.30 and y = 3.0. [e] From heats of formation in the ATcT database, rev. 1.122g (https://atct.anl.gov/).
References
[1] (a) T.H. Dunning, Jr., "Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen", J. Chem. Phys. 1989, 90, 1007. (b) R. A. Kendall, T. H. Dunning, Jr., R. J. Harrison, "Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions", J. Chem. Phys. 1992, 96, 6796. (c) A. K. Wilson, T. van Mourik, T. H. Dunning, Jr., "Gaussian basis sets for use in correlated molecular calculations. VI. Sextuple zeta correlation consistent basis sets for boron through neon", J. Mol. Struct. 1997, 388, 339.
[2] (a) T. Helgaker, W. Klopper, H. Koch, J. Noga, "Basis-set convergence of correlated calculations on water", J. Chem. Phys. 1997 106, 9639. (b) A. Halkier, T. Helgaker, P. Joergensen, W. Klopper, H. Koch, J. Olsen, A. K. Wilson, "Basis-set convergence in correlated calculations on Ne, N2, and H2O", Chem. Phys. Lett. 1998, 286, 243.
[3] S. Zhong, E. C. Barnes, G. A. Petersson, "Uniformly convergent n-tuple-z augmented polarized (nZaP) basis sets for complete basis set extrapolations. I. Self-consistent field energies", J. Chem. Phys. 2008, 129, 184116.
[4] (a) F. Neese, E. F. Valeev, "Revisiting the Atomic Natural Orbital Approach for Basis Sets: Robust Systematic Basis Sets for Explicitly Correlated and Conventional Correlated ab initio Methods?", J. Chem. Theory Comput. 2011, 7, 33. (b) M. Saitow, U. Becker, C. Riplinger, E. F. Valeev, F. Neese, "A new near-linear scaling, efficient and accurate, open-shell domain-based local pair natural orbital coupled cluster singles and doubles theory", J. Chem. Phys. 2016, 146, 164105. (c) A. Altun, F. Neese, G. Bistoni, "Local energy decomposition analysis of hydrogen-bonded dimers within a domain-based pair natural orbital coupled cluster study", Beilstein J. Org. Chem. 2018, 14, 919.
last changes: 12.12.2024, HZ questions & comments to: zipse@cup.uni-muenchen.de