Visualization of Molecular Orbitals using GaussView
GaussView can be used to visualize molecular orbitals in several ways. In the case that cube files for selected orbitals have already been generated for use with other visualization programs, these can also be read with GaussView using the "Open . . " option from the "File" menu. In the popup window giving you an overview of available files, first select "Cube Files" as file type and then select one of the available cube files through mouse click. The contents of the cube file will first appear in a new window in ball-and-stick representation. In order to view the actual MO, select "Surfaces . ." from the "Results" menu. This generates a new window for controlling cube files and surfaces. After the "Cubes Available:" heading you find a scroll list containing all currently loaded/available cubes. This list currently contains only one entry. In order to generate a new surface based on the currently selected cube file, first choose a cutoff isodensity value (0.004 may be appropriate for total SCF densities, values between 0.01 and 0.1 may be appropriate for single orbitals), then select "New Surface" from the "Surfaces Actions ..." menu. This will add a new entry in the list of available surfaces and (after a while) show the surface on top of the ball-and-stick model.
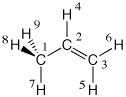
The second approach reads all wavefunction information from the binary checkpoint file (*.chk) or the formatted (textual) checkpoint file (*.fch). The latter can be obtained on addition of the formcheck keyword during a wavefunction calculation with Gaussian. The calculation of molecular orbitals for propene has been chosen here as an example:
#P Becke3LYP/6-31G(d) scf=tight formcheck
Becke3LYP/6-31G(d) propene orbitals with formcheck
0 1
C1
C2 1 r2
C3 2 r3 1 a3
H4 2 r4 3 a4 1 180.0
H5 3 r5 2 a5 1 0.0
H6 3 r6 2 a6 1 180.0
H7 1 r7 2 a7 4 180.0
H8 1 r8 2 a8 7 d8
H9 1 r8 2 a8 7 -d8
r2=1.50202789
r3=1.33329189
r4=1.09120355
r5=1.08850227
r6=1.0867543
r7=1.09519909
r8=1.09844079
a3=125.26675669
a4=118.85584399
a5=121.66815687
a6=121.82450969
a7=111.53888081
a8=111.17619079
d8=120.72760178
|
 |
This run will, among others, generate a checkpoint file in ASCII text format named "Test.FChk", either in the users home or current working directory. It is certainly useful to move the file to a new name after job completion, which is more reflective of the actual system. Since GaussView expects formatted checkpoint files to use the ".fch" ending, a name such as "propene.fch" may be appropriate. In order to read this file with GaussView, select "Open . ." from the "File" menu, select "Gaussian Formatted Checkpoint Files" as the file type, and then select the corresponding file through mouse click. This will open a new window with a ball-and-stick representation of the system. Selecting "Surfaces . ." from the "Results" menu generates a new window for the control of cube files and surfaces. In order to generate pictorial representations of the molecular orbitals, one first has to generate one cube file for each molecular orbital. This is achieved through the appropriate selection in the "Cube Actions ..." menu. One very usefull option is the manual definition of the range of orbitals to be considered. Selecting the option "New Cube" and defining the range of orbitals as "9-16" will, for the case of propene, generate cube files for the highest four occupied and the lowest four unoccupied orbitals.
After generation of the cube files, each of them can be used to generate a 3D representation of one molecular orbital after selecting an isodensity cutoff value and then choosing the "NewSurface" option from the "Surface Action ..." menu. In order to compare a series of molecular orbitals, it is helpful to activate the "New views for new surfaces" button. This will open one new window for each new molecular orbital generated from the cube files.
One particularly useful feature of this approach to viewing the 3D structure of molecular orbitals is that the appearance of the active window can be altered in great detail with the "Display Format ..." option of the "View" menu. In larger systems it is, for example, much more useful to use translucent or mesh representation of the orbitals, which still leave the ball-and-stick model of the system as recognizable background. The "solid" representation type is, of course, most impressive.
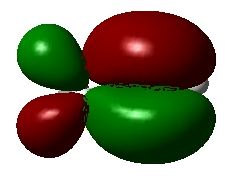
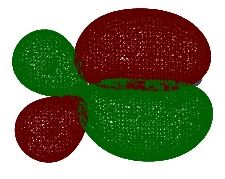
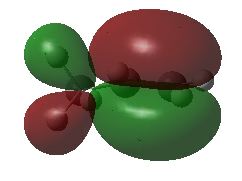
A third approach towards orbital visualization uses the "MOs ..." option of the "Edit" menu in GaussView. This approach is illustrated in great detail in a short text on the Gaussian home page.
Finally, one should always first write down the number of electrons and molecular orbitals as well as the symmetry properties of the system BEFORE starting with the visualization of selected molecular orbitals. What do you expect for propene at the Becke3LYP/6-31G(d) level of theory?